Supervised learning¶
Supervised ML is the task of learning a function that maps an input to an output based on example input-output pairs. Formally, we are given with a set \(\mathcal{D}\) consists of (data,labels) pairs:
where \(x_i \in \mathcal{X}\) are the datapoints and \(y_i \in \mathcal{Y}\) are the labels. For simplicity, we assume here that the “labels space” \(\mathcal{Y}\) is a finite set \(y_i\) that are discrete, univariate variables, i.e., classification settings.
The goal in supervised learning is to fit a function \(f : \mathcal{X} \to \mathcal{Y}\) such that \(f(x_i) =y_i\) for all \(i=1,\dots,m\) . Traditionaly, the data points \(x_i\) are elements of some vector space, meaning that, each point can be expressed using a \(p\)-tuple (vector) of numbers
hence we have \(f : \mathbb{R}^p \to \mathcal{Y}\). However, when working with multi-way data, and time-series data in particular, the situation is different.
The case of matrix valued inputs¶
To adhere with the empirical results and demonstrations shown in 1, we restrict the discussion to the case where data points are gathered across multiple timepoints. That is, each sample \(x_i\) is in fact, an \(n\) by \(p\) matrix, where each of the \(p\) columns represents a feature and the rows correspond to different timepoints in which the samples were gathered.
The matrix input setting is not uncommon, for example, consider the task of gray-scale image classification, where each image is a set of \(n \times p\) pixels and each pixel holds a value from 0 to 1 determining its brightness. In the case of longitudinal sampling data, we can think of the rows of each sample \(x_i\) as discrete samples of a “curve” in a \(p\) dimensional space. In formal words, let \(\gamma_i : \mathbb{R} \to \mathbb{R}^p\) a smooth function, and \(\{t_j\}_{j=1}^n \subset \mathbb{R}\) be a set of timepoints, such that \(\gamma_i (t_j) \in \mathbb{R}^p\) is the \(j^{th}\) row of the matrix \(x_i\) .

An illustration of the \(p\) dimensional curve \(\gamma_i\) (here \(p=3\)), were the blue line is the curve itself, points illustrate discrete sampling process, each point correspond to a timepoint \(t_j\) for \(j=1,\dots ,n\).¶
In our context, we are given with collection of such labeled curves, all of which are sampled in corresponding timepoints, and our goal is to fit a function that takes a sampled curve \(x =[\gamma (t_1), \gamma (t_2) , \dots , \gamma (t_n)]\) and outputs the label associated with the curve.

Illustration of the supervised curve classification task, curves are colored according to their class label.¶
ML workflows¶
The function \(f: \mathbb{R}^{n \times p} \to \mathcal{Y}\) usually consists of a composition of “simpler” functions
In neural networks, \(f_j\) are of the form \(f_j (x) = \alpha_j ({\bf W}_j x + b_j)\) where
\({\bf W}_j\) is a matrix representing a \(\mathbb{R}^{j_1} \to \mathbb{R}^{j_2}\) linear mapping
\(b_j \in \mathbb{R}^{j_2}\) is a bias term
\(\alpha_j\) is a non-linear activation function such as, \(\tanh, \operatorname{SoftMax} , \operatorname{sigmoid}\) etc…
Normalization steps, such as standard-scaling
Dimensionality reduction steps, like PCA, CCA, UMAM, etc…
An essential requirement we impose on the functions \(f_j\) is that after the training process is done, and the parameters are learned, each \(f_j\) must be applicable to samples outside of the training set \(\mathcal{D}\) .
Example
✔ Consider PCA as a dimensionality reduction step, then \(f_j (x) = x {\bf W}_j\) where entries of the matrix \({\bf W}_j\) are determined by the dataset \({\bf X} = [x_1 ; x_2 ; \dots ; x_m]\). Given a new data point \(\bar{x}\), the application of \(f_j\) to \(\bar{x}\) is done by simple matrix multiplication. n
✘ Alternatively, consider the mapping \(f_j\) obtained by fitting a PCoA (or multidimensional scaling) with respect to some dissimilarity funtion. Then the mapping \(f_j\) is relevant only to the samples that are in the training dataset. Applying it as a transformation to samples outside \(\mathcal{D}\) is not straight forward.
In our terminology, functions that are easily applied to data outside of the training dataset are called out-of-sample extendable.
TCAM as dimensionality reduction step¶
The TCAM is a \(\mathbb{R}^{n \times p} \to \mathbb{R}^q\) mapping that is
It makes perfect sense to use it as a dimensionality reduction step within ML workflows.
(*) within the set of pseudo- \(\star_{\bf M}\) orthogonal mappings of rank \(q\) 1
Concrete example¶
We will now construct a concrete example for the incorporation of TCAM in supervised ML workflow for curve classification, using real world microbiome data.
Dataset¶
The data in the following example was obtained from a study by Schirmer et al. 2 . In this study, the authors charactarized the gut microbiome throughout a disease course for 405 pediatric, new-onset, treatment-naive ulcerative colitis (UC) patients.
Patients were monitored for 1 year upon treatment initiation, and microbial taxonomic composition was analyzed from fecal samples using 16S rRNA gene amplicon sequencing. Fecal samples were collected at baseline (week 0, prior to treatment) and 3 follow-up time points (4, 12, and 52 weeks after treatment initiation). At the end of the year, patients were assigned with “label” according their disease state, that is either “Flare” or “Remission”.
Formally:
Inputs: \(\mathcal{X} \subset \mathbb{R}^{n \times p}\) where \(n\) are the four time points, and \(p\) are the number of features obtained by 16S metagenomics sequencing.
Labels: \(\mathcal{Y} = \{ \operatorname{Flare}, \operatorname{Remission} \}\)
We read the data from our git repository as follows
[1]:
import pandas as pd
import numpy as np
data_raw = pd.read_csv("https://raw.githubusercontent.com/UriaMorP/"
"tcam_analysis_notebooks/main/Schirmer2018/Schirmer2018.tsv"
, index_col=[0,1], sep="\t"
, dtype={'Week':int})
labels = pd.read_csv("https://raw.githubusercontent.com/UriaMorP/"
"tcam_analysis_notebooks/main/Schirmer2018/metadata_Schirmer2018.tsv"
, index_col=[0], sep="\t").loc[data_raw.index.get_level_values(0), 'Remission'].rename('label')
labels = 1 * (labels == "Remission") # Binarize the labels
labels = labels.reset_index().copy()
print("Data")
display(data_raw.head(12))
print("Labels")
display(labels.head(4))
Data
| k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Ruminococcaceae-g__Oscillospira-s__-OTU_310886 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Peptostreptococcaceae-g__-s__-OTU_531374 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__-g__-s__-OTU_366584 | k__Bacteria-p__Proteobacteria-c__Betaproteobacteria-o__Burkholderiales-f__Alcaligenaceae-g__Sutterella-s__-OTU_173726 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Ruminococcaceae-g__-s__-OTU_366794 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Lachnospiraceae-g__-s__-OTU_975306 | k__Bacteria-p__Bacteroidetes-c__Bacteroidia-o__Bacteroidales-f__Porphyromonadaceae-g__Parabacteroides-s__distasonis-OTU_585914 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__-g__-s__-OTU_352304 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Lachnospiraceae-g__Blautia-s__-OTU_193061 | k__Bacteria-p__Bacteroidetes-c__Bacteroidia-o__Bacteroidales-f__Prevotellaceae-g__Prevotella-s__-OTU_4302472 | ... | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Lachnospiraceae-g__Roseburia-s__-OTU_313593 | k__Bacteria-p__Synergistetes-c__Synergistia-o__Synergistales-f__-g__-s__-OTU_505549 | k__Bacteria-p__Actinobacteria-c__Coriobacteriia-o__Coriobacteriales-f__Coriobacteriaceae-g__-s__-OTU_4403259 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Lachnospiraceae-g__Epulopiscium-s__-OTU_4371562 | k__Bacteria-p__Bacteroidetes-c__Bacteroidia-o__Bacteroidales-f__S24.7-g__-s__-OTU_1115121 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Ruminococcaceae-g__-s__-OTU_300870 | k__Bacteria-p__Bacteroidetes-c__Bacteroidia-o__Bacteroidales-f__Porphyromonadaceae-g__Parabacteroides-s__distasonis-OTU_186233 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Lachnospiraceae-g__.Ruminococcus-.s__-OTU_300877 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__.Tissierellaceae-.g__-s__-OTU_869796 | k__Bacteria-p__Firmicutes-c__Clostridia-o__Clostridiales-f__Ruminococcaceae-g__Ruminococcus-s__-OTU_196061 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SubjectID | Week | |||||||||||||||||||||
| P_10343 | 0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000869 | 0.022900 | 0.000033 | 0.000000 | 0.0 | ... | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 4 | 0.000183 | 0.000000 | 0.008966 | 0.0 | 0.0 | 0.020421 | 0.000000 | 0.000823 | 0.000000 | 0.0 | ... | 0.000073 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 12 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.006230 | 0.000049 | 0.000997 | 0.000000 | 0.0 | ... | 0.000049 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 52 | 0.000000 | 0.000097 | 0.000000 | 0.0 | 0.0 | 0.002211 | 0.000000 | 0.000291 | 0.000000 | 0.0 | ... | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000048 | 0.0 | 0.0 | |
| P_10897 | 0 | 0.000423 | 0.000000 | 0.000059 | 0.0 | 0.0 | 0.077260 | 0.000863 | 0.017448 | 0.000051 | 0.0 | ... | 0.000008 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 4 | 0.000023 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.006095 | 0.000411 | 0.000000 | 0.000000 | 0.0 | ... | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 12 | 0.000080 | 0.000013 | 0.010809 | 0.0 | 0.0 | 0.005411 | 0.000372 | 0.000359 | 0.000027 | 0.0 | ... | 0.000027 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 52 | 0.000000 | 0.000080 | 0.000664 | 0.0 | 0.0 | 0.013934 | 0.000261 | 0.000905 | 0.000000 | 0.0 | ... | 0.000040 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| P_1108 | 0 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.002086 | 0.000000 | 0.000066 | 0.000016 | 0.0 | ... | 0.000049 | 0.000000 | 0.0 | 0.0 | 0.000049 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 4 | 0.000000 | 0.000211 | 0.000000 | 0.0 | 0.0 | 0.011007 | 0.000000 | 0.000409 | 0.000000 | 0.0 | ... | 0.000384 | 0.000025 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 12 | 0.000007 | 0.000000 | 0.000028 | 0.0 | 0.0 | 0.038760 | 0.000528 | 0.003105 | 0.000000 | 0.0 | ... | 0.000428 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | |
| 52 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.115325 | 0.002798 | 0.018651 | 0.000000 | 0.0 | ... | 0.000311 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
12 rows × 1015 columns
Labels
| SubjectID | label | |
|---|---|---|
| 0 | P_10343 | 0 |
| 1 | P_10343 | 0 |
| 2 | P_10343 | 0 |
| 3 | P_10343 | 0 |
Making the ML workflow¶
We rely on sklearn’s Pipeline interface in order to implement the composition of functions, previously formulated as \(f = f_1 \circ f_2 \circ ... \circ f_d\) making the classification model.
A python code realization of the above mathematical formulation would be as follows:
from sklearn.pipeline import Pipeline
pipe = Pipeline([
('f_1', Transformer1())
,('f_2', Transformer2())
# ...
,('f_d', Estimator())
])
where Transformers are classes implementing sklearn.base.TransformerMixin and Estimator implements sklearn.base.BaseEstimator .
Filtration of low-abundant features
Curvewise standardization
TCAM truncation
MinMax scaling
GradientBoostClassifier
For steps 1.-3. it is most convenient to work with pandas.DataFrames as inputs.
[2]:
from sklearn.base import BaseEstimator, TransformerMixin
# step 1: Filtration of low-abundant features
class RAFilter(BaseEstimator, TransformerMixin):
def __init__(self, capval = 5e-2):
self._f_list = []
self.capval = capval
pass
def fit(self, X,y=None, modemap = []):
X_t = X.copy()
X_t = 100 * (X_t / X_t.values.sum(axis=1,keepdims = True))
self._f_list = [i for i in range(X_t.shape[1]) if (X_t.iloc[:,i] > self.capval).sum() > X_t.shape[0]*0.1 ]
return self
def transform(self, X, y = None):
X_t = 100 * (X / X.values.sum(axis=1,keepdims = True)).copy()
X_t = X_t.iloc[:,self._f_list]
X_t[X_t < self.capval] = self.capval
X_t = 100 * X_t / X_t.values.sum(axis=1,keepdims = True)
return X_t
def fit_transform(self, X, y = None, modemap = []):
self = self.fit(X, y)
Xt = self.transform(X, y)
return Xt
Since microbiome data tends to cluster by participant, meaning that variation coming from interindividual differences tend overshadow any other source of variation, including temporal trends, the task of identifying mutual temporal trajectories in microbiome composition is very challenging. Keeping in mind that we are only concerned with the direction of temporal variation trends (rather than the concrete baseline), one simple way of tackling this challenge would be to look at deviations from the baseline period. That is, instead of looking at raw values \(x_{ij}\) (the \(j^{th}\) time point of subject \(i\)), we consider tthe followoing:
where \(\mu ( x_{i0} )\) denotes the baseline sample of subject \(i\) . Using pandas, this transfromation looks like:
[3]:
from sklearn.preprocessing import FunctionTransformer
# step 2: Curvewise standardization
def standardize_curve_df(X, y = None, cap = 5e-3):
blmean = (X + cap).groupby(level=['SubjectID']).apply(lambda x:x / x.loc[x.index.get_level_values('Week') == 0].iloc[0] )
blmean = np.log2(blmean)
return blmean
# Wrap the function with Transformer
StandardCurve = FunctionTransformer(standardize_curve_df)
Step 3. is the last step applied to a dataframe. In this step, we cast the dataframe into a tensor, then a TCAM is applied, followed by truncation (dimensionality reduction step) and a numpy.array is returned.
Gotcha
Suppose that the size of the training dataset is \(4m\) in its tabular form, the number of rows in the output of TCAMWrapper.transform is \(m\)
For many good reasons, this reduction in number of samples , is not allowed by the Pipeline (it throws a ValueError: Found input variables with inconsistent numbers of samples: [m, 4m] )
In our case, this decrease is intentional, and we hack our way around sklearn’s safeguards
To overcome this inconsistency, we divide the workflow into two pipelines each of which maintains consistent number of rows, and the TCAM transformation is patched between the two pipelines.
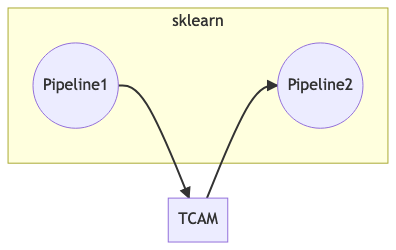
The pipeline patching process, the inputs and outputs of Pipeline1 all have \(4m\) rows, while those of Pipeline1 have \(m\) rows.
The TCAM transformation (of matrix to tensors) takes inputs with \(4m\) rows and outputs tables of \(m\) rows.¶
[4]:
from sklearn.pipeline import Pipeline
from sklearn.preprocessing import MinMaxScaler
from sklearn.neural_network import MLPClassifier
from sklearn.ensemble import RandomForestClassifier, GradientBoostingClassifier
from mprod.dimensionality_reduction import TCAM
from mprod import table2tensor
class Patch(BaseEstimator, TransformerMixin):
def __init__(self,pipeline1, tcam_obj):
self.pipeline1 = pipeline1
self.tcam_obj = tcam_obj
def _transform_y(self, y, map1):
yt = pd.Series(map1).rename('row').to_frame()
yt = yt.merge(y.reset_index(), left_index = True, right_on = "SubjectID").drop_duplicates()
yt.index = yt['row']
yt = yt['label'].sort_index()
return yt
def fit(self, X, y = None):
self.pipeline1 = self.pipeline1.fit(X,y=y)
tensor, map1, map3 = table2tensor(self.pipeline1.transform(X))
if y is not None:
yt = self._transform_y(y,map1)
else:
yt = None
self.tcam_obj = self.tcam_obj.fit(tensor,y=yt)
return self
def transform(self, X):
Xt1 = self.pipeline1.transform(X)
tensor, map1, map3 = table2tensor(Xt1)
Xt2 = self.tcam_obj.transform(tensor)
return Xt2
Instantiation on this workflow is done as follows:
[5]:
rs = 0 # To make sure executions are consistant
pipeline1 = Pipeline([
("raf", RAFilter(capval = 5e-3)),
("st_curve", StandardCurve)
])
pipeline2 = Pipeline([
('mmt',MinMaxScaler(clip = True)),
('clf', GradientBoostingClassifier(learning_rate=.2,random_state=rs))
])
patch = Pipeline([
('pipeline1',Patch(pipeline1, TCAM(n_components=.8)))
, ('pipeline2', pipeline2)])
Fron here and on, the patch object is just like any other classification model, and can be evaluated as such
Model evaluation¶
[6]:
from sklearn.model_selection import StratifiedKFold
from sklearn.metrics import auc
from sklearn.metrics import plot_roc_curve, roc_curve
import matplotlib.pyplot as plt
%matplotlib inline
cv = StratifiedKFold(n_splits=6, shuffle=True, random_state=rs)
# indices for CV iterations
sid_ser = labels.copy().drop_duplicates()
sid_ser.index = sid_ser['SubjectID']
sid_ser = sid_ser['label']
dummy_size = np.arange(sid_ser.values.size)
# storing the model's scores
tprs = []
aucs = []
mean_fpr = np.linspace(0, 1, 100)
# CV loop
fig, ax = plt.subplots()
for i, (train_iloc, test_iloc) in enumerate(cv.split(dummy_size,sid_ser.values, sid_ser.values)):
train_id = sid_ser.index[train_iloc].copy().sort_values()
test_id = sid_ser.index[test_iloc].copy().sort_values()
y_train = sid_ser[train_id].copy()
X_train = data_raw.loc[data_raw.index.get_level_values("SubjectID").isin(y_train.index)].copy().sort_index(0)
y_test = sid_ser[test_id].copy()
X_test = data_raw.loc[data_raw.index.get_level_values("SubjectID").isin(y_test.index)].copy().sort_index(0)
trained_model = patch.fit(X_train, y_train.copy())
viz = plot_roc_curve(trained_model, X_test, y_test.copy(),
name='F{}'.format(i+1),
alpha=0.3, lw=1, ax=ax)
interp_tpr = np.interp(mean_fpr, viz.fpr, viz.tpr)
interp_tpr[0] = 0.0
tprs.append(interp_tpr)
aucs.append(viz.roc_auc)
ax.plot([0, 1], [0, 1], linestyle="--", lw=2, color="r", label="Chance", alpha=0.8)
mean_tpr = np.mean(tprs, axis=0)
mean_tpr[-1] = 1.0
mean_auc = auc(mean_fpr, mean_tpr)
std_auc = np.std(aucs)
ax.plot(
mean_fpr,
mean_tpr,
color="b",
label=r"Mean AUROC = %0.2f " % (mean_auc),
lw=2,
alpha=0.8,
)
std_tpr = np.std(tprs, axis=0)
tprs_upper = np.minimum(mean_tpr + std_tpr, 1)
tprs_lower = np.maximum(mean_tpr - std_tpr, 0)
ax.fill_between(
mean_fpr,
tprs_lower,
tprs_upper,
color="grey",
alpha=0.2,
label=r"$\pm$ 1 std. dev.",
)
ax.set(
xlim=[-0.05, 1.05],
ylim=[-0.05, 1.05],
)
ax.legend(loc=[.6, 0.05], fontsize = 6 )
ax.set_ylabel("TPR")
ax.set_xlabel("FPR\n(positive label: 1)")
fig.set_size_inches([2.1,2.1])
fig.set_dpi(300)
plt.show()
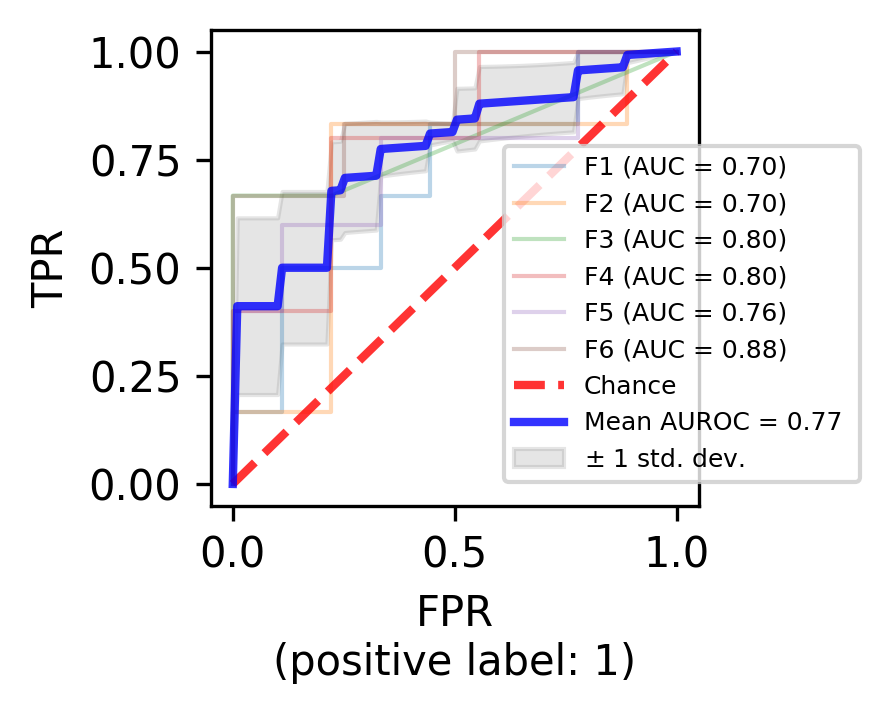
- 1(1,2)
Uria Mor, Yotam Cohen, Rafael Valdes-Mas, Denise Kviatcovsky, Eran Elinav, and Haim Avron. Dimensionality reduction of longitudinal ‘omics data using modern tensor factorization. 2021. arXiv:2111.14159.
- 2
Melanie Schirmer, Lee Denson, Hera Vlamakis, Eric A. Franzosa, Sonia Thomas, Nathan M. Gotman, Paul Rufo, Susan S. Baker, Cary Sauer, James Markowitz, Marian Pfefferkorn, Maria Oliva-Hemker, Joel Rosh, Anthony Otley, Brendan Boyle, David Mack, Robert Baldassano, David Keljo, Neal LeLeiko, Melvin Heyman, Anne Griffiths, Ashish S. Patel, Joshua Noe, Subra Kugathasan, Thomas Walters, Curtis Huttenhower, Jeffrey Hyams, and Ramnik J. Xavier. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host & Microbe, 24(4):600–610.e4, oct 2018. URL: https://linkinghub.elsevier.com/retrieve/pii/S1931312818304931, doi:10.1016/j.chom.2018.09.009.